LE PROJET POPGEN Aider au diagnostic génétique des maladies rares en séquençant la population française
Kevin Uguen, Praticien Hospitalier Universitaire
Gaëlle Le Folgoc, Cheffe de projet
Emmanuelle Génin, Directrice de Recherche InsermCHU de Brest et UMR1078 Génétique, Génomique fonctionnelle et Biotechnologies, UBO/Inserm/EFS
Le monde des maladies rares, qui touchent plus de trois millions de patients en France, a connu une révolution ces dernières années avec l’arrivée des nouvelles technologies de séquençage, qui nous permettent d’analyser la totalité de notre génome en quelques jours. Pourtant, identifier parmi les quelques millions de variations de séquence que comporte le génome d’un patient celle qui est responsable de sa maladie peut s’avérer difficile. Pour cela, il faut comparer ces variations à celles retrouvées dans une population témoin. C’est l’objectif du projet POPGEN, qui a permis de séquencer 4 000 individus français afin de créer une base de données au service du soin pour interpréter le génome des patients.
Les maladies rares touchent chacune moins de 1 personne sur 2 000 mais du fait de leur multiplicité, on en dénombre plus de 7 000, elles représentent un réel problème de santé publique avec plus de trois millions de patients en France et plus de 300 millions dans le monde. Ce sont des maladies généralement sévères qui peuvent toucher différents organes, surviennent pour une grande partie d’entre elles dès la naissance ou dans l’enfance et ont une cause génétique dans 80 % des cas. Ces maladies sont souvent mal diagnostiquées car, en cause de leur rareté, elles restent encore mal connues et les patients n’obtiennent un diagnostic que très tardivement, quand ils l’obtiennent. Ces dernières années cependant, des progrès considérables ont été obtenus grâce aux nouvelles techniques de séquençage du génome qui permettent d’obtenir un diagnostic dans 50 % des cas contre seulement 20 % il y a encore une dizaine d’années.
Le plan France médecine génomique 2025 : une égalité d’accès au séquençage du génome
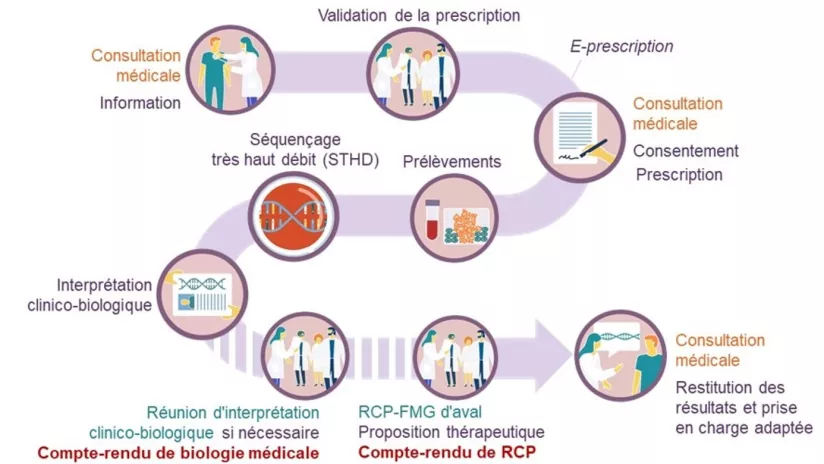
Le séquençage du génome humain a connu, au cours des dernières décades, une véritable révolution. Alors que le premier projet de séquençage, le « human genome project » (HGP), s’est étendu de 1990 à 2003 et a coûté 3 milliards de dollars1, il est aujourd’hui possible de séquencer la totalité du génome humain en quelques heures et pour quelques centaines d’euros. Cette diminution drastique des coûts et des temps d’analyse a permis la démocratisation de cette technique, qui équipe de nos jours de nombreux CHU ainsi que des établissements de recherche. Pour aller encore plus loin dans cette voie, la France s’est dotée en 2016 d’un plan national, le plan France médecine génomique 2025 (PFMG2025). Le PFMG2025 a permis la mise en place de deux laboratoires de séquençage très haut débit, qui permettent d’analyser le génome complet de plusieurs milliers de patients atteints de maladies rares et cancers de tout le territoire français2. Un parcours précis a été mis en place, centré sur le patient, où chaque situation est discutée en réunion de concertation pluridisciplinaire, en amont et en aval de l’analyse (Figure 1).
Explorer les zones d’ombre du génome pour améliorer le rendement diagnostique
Malgré les améliorations apportées par le séquençage, il persiste une impasse diagnostique pour la moitié des patients atteints de maladie rare, et une errance diagnostique de plus de quatre ans pour un quart des malades, ce qui a un impact négatif sur leur prise en charge. Ces difficultés de diagnostic ne s’expliquent pas par le fait que la cause de la maladie n’est pas dans le génome du patient mais, dans de nombreux cas, par notre mauvaise connaissance de l’organisation du génome et des rôles joués par les différents éléments qui le constituent. En effet, être capable techniquement de séquencer les trois milliards de paires de base que contiennent notre génome ne suffit pas pour rendre un diagnostic, il faut encore pouvoir interpréter les variations qui sont observées chez le patient pour identifier celle(s) qui est(sont) responsable(s) de sa maladie. Et c’est bien là que se situe le défi car dans le génome d’un individu, on observe plus de trois millions de variations et ces variations peuvent prendre différentes formes et se situer dans différentes parties du génome.
La majorité des variations aujourd’hui connues pour être impliquées en pathologie concernent les régions codantes du génome, c’est à dire les régions des gènes codant une protéine. C’est d’ailleurs vers ces variations que le biologiste concentre son effort lors d’une analyse de génome (Figure 2). Or notre génome est constitué à 98 % de séquences dites non-codantes. Ces séquences, initialement décrites comme de l’ADN « poubelle », restent encore très mal connues mais on soupçonne qu’elles jouent un rôle très important pour le bon fonctionnement de nos cellules, en participant par exemple à la régulation de l’expression des gènes3. Récemment un exemple frappant est venu illustrer l’importance de ces régions : une variation récurrente dans RNU4-2, un petit ARN non codant, a été décrite comme une des causes génétiques les plus fréquentes de troubles du neurodéveloppement4. Jusqu'à présent, cette variation était restée non identifiée, car elle se situe dans une région peu explorée. Ce n’est qu’avec la démocratisation du séquençage du génome et à la constitution de cohortes de patients et de témoins que cette nouvelle cause de déficience intellectuelle a pu être révélée, offrant ainsi une issue à l’impasse diagnostique pour plusieurs centaines de patients à travers le monde.
De l’importance de bien connaître la variabilité génétique de la population
Pour interpréter le rôle joué par les différentes parties du génome et l’impact que peuvent avoir les variations génétiques observées chez un patient, il est essentiel de pouvoir comparer ces variations à celles observées dans la population dont le patient est issu. L’idée est de voir si ces variations sont également présentes chez d’autres individus de la même population qui ne sont pas malades pour pouvoir les ignorer et se concentrer sur celles, plus rares, et plus susceptibles d’être impliquées dans la maladie. Cela semble simple mais encore faut-il disposer de données sur la fréquence des variations dans une population témoin qui représente bien la population d’origine du patient. Et c’est là que les choses se compliquent…
En effet, s’il existe différentes bases de données qui répertorient les variations observées dans différentes populations à travers le monde, principalement d’Europe du Nord, mais aussi d’Afrique et d’Asie, ces informations ne sont pas suffisamment précises pour pouvoir savoir si une variation observée chez un patient originaire d’une région française donnée existe et est fréquente dans cette région précise. On a en effet montré que des différences régionales existent à l’intérieur de la France comme d’ailleurs dans la majorité des pays étudiés jusqu’à présent, différences qui sont le reflet de l’histoire démographique des populations5,6.
Le projet POPGEN, un panel de référence pour aider à interpréter les variations présentes chez les patients
Devant ce besoin de couverture du territoire national, le PFMG2025 a initié un projet pilote en population générale pour doter les soignants et les chercheurs d’un « outil » aidant à l’interprétation des variants observés dans le génome de patients ; c’est la naissance du projet POPGEN.
Pour ce projet, nous avons travaillé en collaboration avec l’équipe de coordination de la cohorte Constances (Encadré 1) pour envoyer à l’ensemble des volontaires un questionnaire sur les lieux de naissance de leurs parents et grands-parents, questionnaire que nous avons pu inclure dans le questionnaire de suivi annuel de 2019. Nous avons ensuite sélectionné 15 000 de ces volontaires sur la base de ces lieux de naissance en privilégiant les volontaires dont les 4 grands-parents étaient nés dans une même zone géographique en France tout en cherchant à maximiser la couverture nationale. Ces 15 000 volontaires ont alors reçu une enveloppe contenant un kit d’auto-prélèvement salivaire, une notice d’information ainsi qu’un formulaire de consentement, leur proposant de participer à la recherche POPGEN qui est une recherche impliquant la personne humaine (RIPH) de type 37. Au premier trimestre 2022, 10 250 volontaires avaient consenti et donc renvoyé un échantillon salivaire accompagné d’un formulaire de consentement signé. Les ADN ont été extraits des échantillons salivaires à la Fondation Jean Dausset - Centre d’Étude du Polymorphisme Humain (CEPH) puis un génotypage a été réalisé au Centre National de Recherche en Génomique Humaine (CNRGH). Le génotypage consiste à analyser uniquement une fraction du génome pour vérifier la qualité du prélèvement et obtenir une première partie des informations génétiques indispensables pour la suite du projet. Au total, nous avons pu obtenir des données de génotypage exploitables pour 9 772 volontaires, et parmi eux, sélectionner 4 000 individus pour un séquençage complet du génome. Ce séquençage a été finalisé au premier trimestre 2024 et les données sont en cours d’analyse pour fournir à terme une base de données de référence des variations génétiques et de leurs fréquences dans les différentes régions de France métropolitaine. Cette base de données vise à s’enrichir également de données issues d’individus originaires des territoires ultra-marins ainsi que des résidents français, sans distinction de leur origine géographique. Elle s’intégrera également dans le projet européen « Genome Of Europe », dont l’objectif est de réaliser ce même type d’étude à l’échelle de toute l’Europe et de promouvoir le partage des données dans un environnement sécurisé et respectueux des participants8.
La cohorte Constances
La cohorte Constances (Consultants des Centres d’Examens de Santé)9, d’où sont sélectionnés les individus du projet POPGEN, est la plus grande cohorte épidémiologique en population française. C’est une infrastructure nationale de recherche en biologie et en santé financée dans le cadre de Programme des Investissement d’Avenir (PIA). S’intéressant aux causes des maladies, notamment multifactorielles, elle consiste à suivre individuellement et sur plusieurs années un groupe d’individus, en recueillant entre autres des données sur l’exposition à des facteurs de risque de maladies, sur la survenue de pathologies... Elle est composée de 220 000 volontaires majeurs nés entre 1941 et 2000, recrutés entre 2012 et 2021. Les individus ayant accepté l’inclusion ont bénéficié d’examens médicaux, ont rempli divers questionnaires et des échantillons de sang et d’urines sont recueillis régulièrement. Cette cohorte est ouverte à la communauté de recherche, et tout projet proposé par une équipe de recherche est évalué par un conseil scientifique international.
Réutilisation des données du diagnostic pour la recherche
La mise en place des laboratoires de séquençage SeqOia et Auragen permet la génération de données de séquence de plusieurs milliers de patients dans un cadre diagnostique sur des pré-indications précises. Au travers du CAD (Collecteur Analyseur de Données), qui se met en place au sein du PFMG2025, les chercheurs pourront, si le comité scientifique et éthique a accepté le projet et en l’absence d’opposition du patient, réanalyser les données de séquençage des patients afin, par exemple, de tenter d’identifier de nouveaux gènes responsables de pathologies. Les données générées dans le cadre du projet POPGEN seront également accessibles via le CAD pour permettre de facilement exclure des génomes de patients les variations fréquentes dans la population générale et accélérer le diagnostic. Ces données pourront également permettre de nouvelles études sur la structure génétique de la population pour mieux comprendre le rôle joué par les gènes et leurs variations dans les maladies et la santé.
- 1. International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431(7011):931-945. doi:10.1038/nature03001
- 2. https://pfmg2025.aviesan.fr/le-plan/lbm-fmg
- 3. ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306(5696):636-640. doi:10.1126/science.1105136
- 4. Chen Y, Dawes R, Kim HC, et al. De novo variants in the RNU4-2 snRNA cause a frequent neurodevelopmental syndrome. Nature. 2024;632(8026):832-840. doi:10.1038/s41586-024-07773-7
- 5. Saint Pierre A, Giemza J, Alves I, et al. The genetic history of France [published correction appears in Eur J Hum Genet. 2020 Jul;28(7):988. doi: 10.1038/s41431-020-0604-1]. Eur J Hum Genet. 2020;28(7):853-865. doi:10.1038/s41431-020-0584-1
- 6. Alves I, Giemza J, Blum MGB, et al. Human genetic structure in Northwest France provides new insights into West European historical demography. Nat Commun. 2024;15(1):6710. Published 2024 Aug 7. doi:10.1038/s41467-024-51087-1
- 7. https://lysine.univ-brest.fr/popgen
- 8. https://b1mg-project.eu/1mg/genome-europe
9. Zins M, Goldberg M; CONSTANCES team. The French CONSTANCES population-based cohort: design, inclusion and follow-up. Eur J Epidemiol. 2015;30(12):1317-1328. doi:10.1007/s10654-015-0096-4